Our research focuses on using genomic data from both ancient and present-day individuals to reconstruct human evolutionary history and understand the forces shaping genetic variation. By integrating large-scale datasets with new analytical approaches, we aim to understand: (1) how evolutionary processes such as mutation rate evolve across individuals, (2) reconstruct key evolutionary events––such as gene flow, population bottlenecks and expansions–– in human history, and (3) leverage population history to identify genetic variants related to human adaptation and disease. Some ongoing projects in the lab are described below.
Reconstructing population history using genetic data
We develop and apply computational methods to reconstruct human evolutionary history using genomic data from present-day and ancient individuals. Our work focuses on inferring population relationships, detecting and dating admixture events, and identifying deeply divergent “ghost” ancestry that is not represented in sampled genomes. By leveraging patterns of genetic variation and introgressed segments, we uncover complex demographic histories, including multiple waves of archaic admixture and population structure across space and time. These approaches enable us to move beyond simplified models and provide a more detailed and quantitative understanding of human origins and migrations.
Image: Meaghan Marohn
Representative lab publications on this topic include:
- Zhang*,†, Biddanda A*,†, Johnson SA, Dushlaine CO, Moorjani P†. Recovering signatures of archaic introgression using ancestral recombination graphs. bioRxiv 2026.03.03.709416.
- Iasi LM*,†, Chintalapati M*,†, Skov L , Mesa AB , Hajdinjak M, Peter BM*,†, Moorjani P*,†. Neandertal ancestry through time: Insights from genomes of ancient and present-day humans. Science 386.6727 (2024): eadq3010.
- Kerdoncuff E*,†, Skov L*,†, Patterson N, Banerjee J, Khobragade P, Chakrabarti SS, Chakrawarty A, Chatterjee P, Dhar M, Gupta M, John JP, Koul PA, Lehl SS, Mohanty RR, Padmaja M, Perianayagam A 12, Rajguru C, Sankhe L, Talukdar A, Varghese M, Yadati SR, Zhao W, Leung YY, Schellenberg GD, Wang YZ, Smith JA, Dey S, Ganna A , Dey AB†, Kardia SLR†, Lee J†, Moorjani P†. 50,000 years of Evolutionary History of India: Impact on Health and Disease Variation. Cell 188 (2025):13, 3389-3404.e6.
- Chintalapati M+, Patterson N+, Moorjani P+. (2022) Reconstructing the spatiotemporal patterns of admixture during the European Holocene using a novel genomic dating method. eLife. DOI: https://doi.org/10.7554/eLife.77625.
- Tournebize R+, Chu G and Moorjani P+. (2022). Reconstructing the history of founder events using genome-wide patterns of allele sharing across individuals. PLoS Genetics. https://doi.org/10.1371/journal.pgen.1010243.
- Moorjani P, Sankararaman S, Fu Q, Przeworski M, Patterson N, Reich D. (2016). A genetic method for dating ancient genomes provides a direct estimate of human generation interval in the last 45,000 years. Proc Natl Acad Sci USA. May 17;113(20):5652-7. doi: 10.1073/pnas.1514696113.
Leveraging evolutionary history for identifying disease and adaptive variants
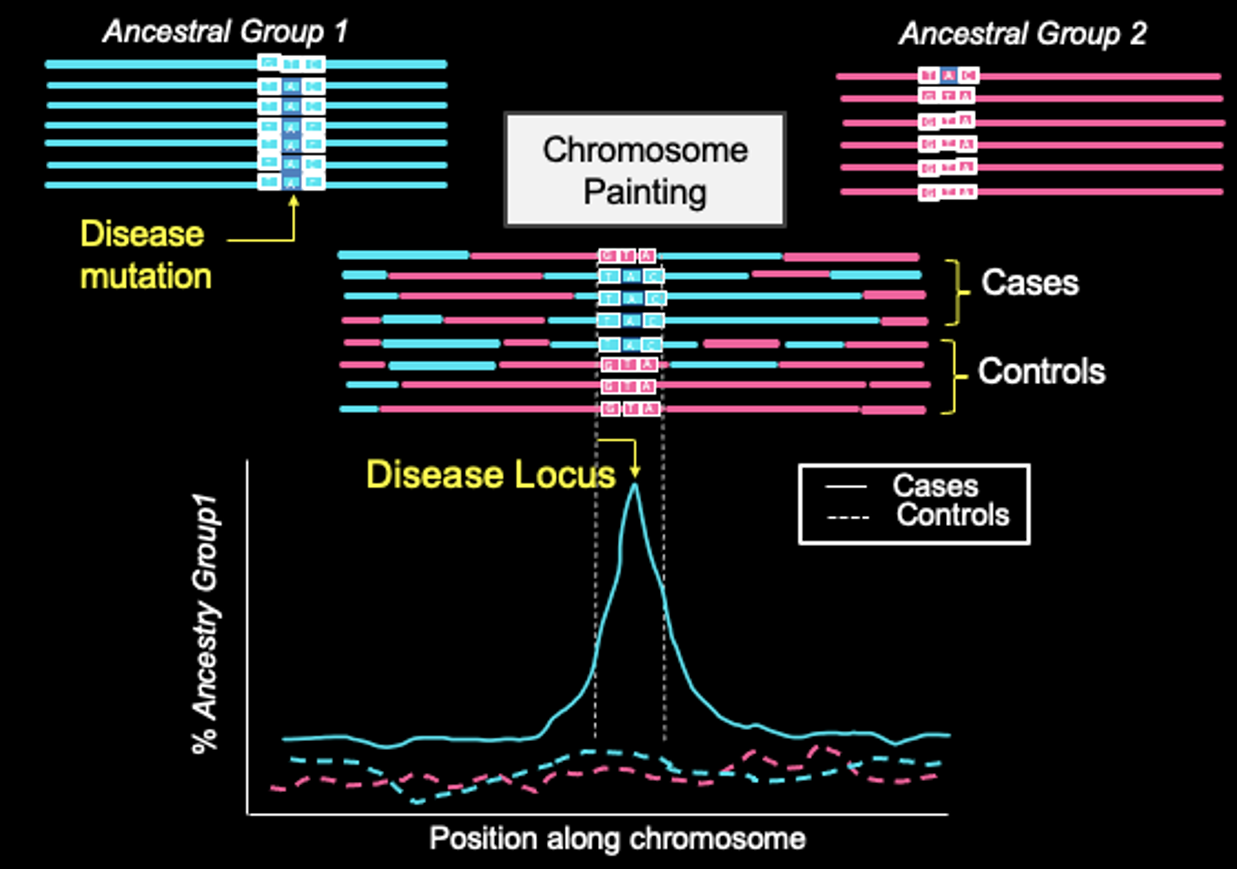
Image: Manjusha Chintalapati
We use insights from human evolutionary history to identify genetic variants that influence adaptation and disease risk. By studying patterns of natural selection, archaic introgression, and population-specific genetic variation, we aim to pinpoint variants that have functional and phenotypic consequences. Our work leverages large-scale genomic datasets to understand how evolutionary forces—such as admixture, founder events and demographic history—shape the distribution of disease-associated alleles across populations. As one example, we are developing methods such as admixture mapping (illustrated in the figure), which uses local ancestry variation to localize disease-associated loci by identifying regions with excess ancestry from higher-risk populations. Together, these approaches provide a powerful framework for interpreting genetic variation in biomedical studies, particularly in underrepresented populations.
Representative lab publications on this topic include:
- Kerdoncuff E*, Marohn M*, Cramer N, Dey S, Kardia S, Thangaraj K, Ségurel L, Lee J, Dey AB, Moorjani P. Revisiting the Evolution of Lactase Persistence: Insights from South Asian Genomes. bioRxiv 2025.11.05.686799.
- Kerdoncuff E*,†, Skov L*,†, Patterson N, Banerjee J, Khobragade P, Chakrabarti SS, Chakrawarty A, Chatterjee P, Dhar M, Gupta M, John JP, Koul PA, Lehl SS, Mohanty RR, Padmaja M, Perianayagam A 12, Rajguru C, Sankhe L, Talukdar A, Varghese M, Yadati SR, Zhao W, Leung YY, Schellenberg GD, Wang YZ, Smith JA, Dey S, Ganna A , Dey AB†, Kardia SLR†, Lee J†, Moorjani P†. 50,000 years of Evolutionary History of India: Impact on Health and Disease Variation. Cell 188 (2025):13, 3389-3404.e6.
- , , , , , , , , Effect of APOE e4 and its modification by sociodemographic characteristics on cognitive measures in South Asians from LASI-DAD. Alzheimer’s Disease & Dementia.
- Moorjani P, Thangaraj K, Patterson N, Lipson M, Loh PR, Govindaraj P, Berger B, Reich D, Singh L. Genetic evidence for recent population mixture in India. (2013). American Journal of Human Genetics. Sep 5;93(3):422-38. doi: 10.1016/j.ajhg.2013.07.006.
- , , , , ,, , , , , , , , , , , K. (2017). The promise of disease gene discovery in South Asia. Nature Genetics, doi:10.1038/ng.3917.
Evolution of mutation rate and spectrum across individuals
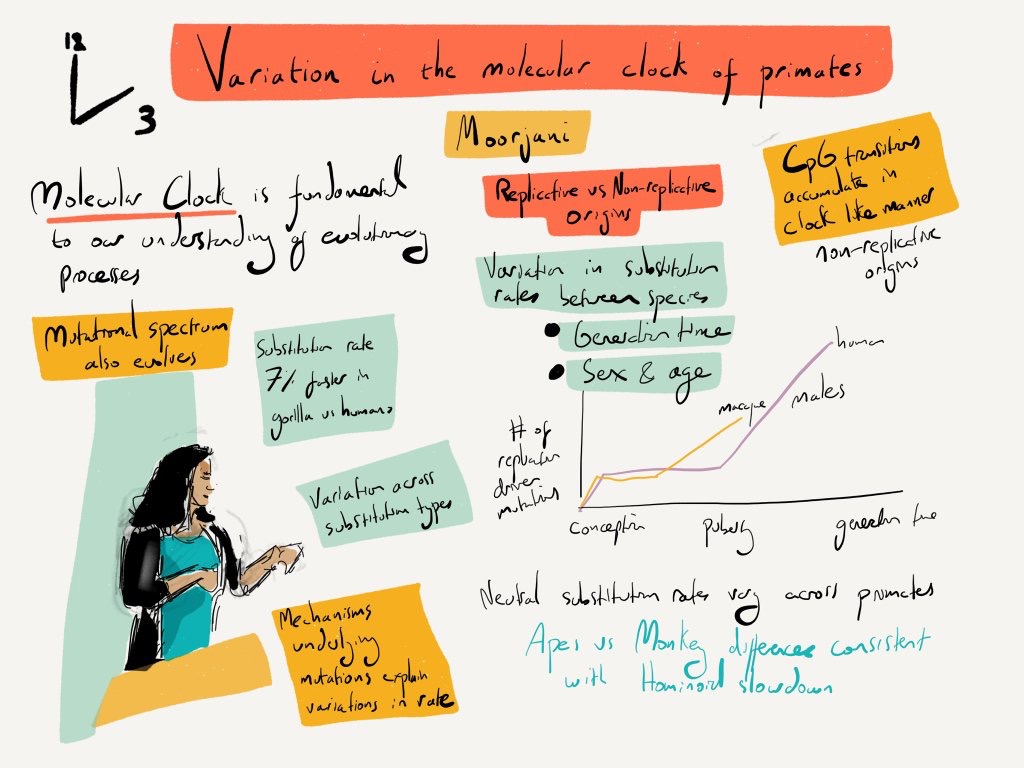
Image: Alex Cagan, CSHL 2016
We investigate how mutation rates vary across the genome, across populations, and over evolutionary timescales. Our work develops statistical methods to characterize fine-scale variation in mutation processes, identify mutational hotspots, and understand how genomic features and evolutionary forces shape mutation rate heterogeneity. By comparing patterns across species and time, we aim to uncover the mechanisms driving mutation rate evolution and their consequences for genetic diversity, adaptation, and disease. This work provides fundamental insights into one of the key processes generating genetic variation.
Representative lab publications on this topic include:
- Gao Z+, Zhang Y, Cramer N, Przeworski M, Moorjani P+ (2022). Limited role of generation time changes in driving the evolution of the mutation spectrum in humans. eLife 12, e81188. DOI: https://doi.org/10.7554/eLife.81188.
- , , , , , A comparison of humans and baboons suggests germline mutation rates do not track cell divisions. PLoS biology. 18, e3000838.
- Chintalapati M and Moorjani P+. (2020). Evolution of the mutation rate across primates. Current Opinion in Genetics & Development. 62, 58-64.
- Moorjani P, Gao Z, and Przeworski M. (2016). Human germline mutation and the erratic evolutionary clock. PLoS Biol 14(10): e2000744. doi:10.1371/journal.pbio.2000744.
- Moorjani P , Amorim CE, Arndt P, and Przeworski M. (2016). Variation in the molecular clock in primates. Proc Natl Acad Sci USA. Sep 6. pii: 201600374.
